

Спиналната мускулна атрофия е рядко и често фатално генетично заболяване, което води до мускулна слабост и прогресивна загуба на двигателна функция.
То засяга 1 на 11 000 новородени. В България са диагностицирани около 160 пациенти с това рядко заболяване, като в момента те имат достъп до лечение по всички най-нови стандарти.
В зависимост от възрастта при изява на първите симптоми и тежестта на протичане заболяването се класифицира в пет типа – 0, 1, 2, 3, 4.
Спиналната мускулна атрофия има различни форми, като най-тежката е тип 1, която обикновено се проявява в ранна детска възраст.
Какво представлява спиналната мускулна атрофия?
Спиналната мускулна атрофия (СМА) е генетично невромускулно заболяване, при което специализирани нервни клетки в гръбначния мозък и мозъчния ствол постепенно загиват.
„При недостиг на протеина SMN тези неврони се повреждат, а мускулите отслабват прогресивно“, обясняват от Националния институт по неврологични заболявания и инсулт (NINDS).
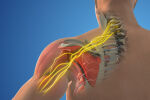
Генетичната основа
Коренът на проблема е мутация в гена SMN1. Тежестта на клиничната картина зависи от броя копия на „резервния“ ген SMN2 – колкото повече са те, толкова по-леко протича болестта. Наследяването е автозомно-рецесивно: при двама носители рискът всяко дете да бъде засегнато е 25%.
Степени на Спинална мускулна атрофия
СМА обикновено се категоризира в няколко типа въз основа на възрастта, на начало и тежестта на симптомите:
Тип 0 (пренатален)
Това е най-рядката и най-сериозна форма на СМА. Тип 0 често започва, когато бебето расте в утробата.

Бебето може:
- Да се движи по-рядко или да спре да се движи в утробата
- Да се роди със сериозна мускулна слабост
- Да има по-меки, отпуснати мускули (хипотония)
- Да липсват рефлекси
- Да има парализа от двете страни на лицето
- Да има сърдечни проблеми при раждането (вродени сърдечни проблеми)
- Да има слаби дихателни мускули, пише webmd.
Тип 1 (най-тежката форма, започваща в ранна детска възраст)
Може да се появи между раждането и 6-месечна възраст. Много деца с тип 1 имат два до три SMN2 гена. Въпреки че мускулите на бебето може да са силни в началото, те отслабват с времето. Синдромът засяга главно долните крайници. Бебе може:
- Да не може да поддържа главата си самостоятелно
- Да не седи изправено без помощ
- Да има отпуснати ръце и крака (хипотония)
- Да има проблеми с преглъщането или по-висок риск от поглъщане на течност в дихателните си пътища
- Да движи езика си неравномерно
- Да събира слюнка
- Да има проблеми с дишането
- Да има камбановиден гръден кош
- Да се развива по-бавно
- Да губи тегло или да не яде толкова много
Някои видове лечения могат да помогнат за доставянето на кислород до тъканите и органите на вашето бебе. Поради проблеми с дишането много деца живеят до около 2-годишна възраст. Но с респираторно лечение някои деца могат да живеят по-дълго.
Тип 2 (хронична форма, започваща в ранна детска възраст)
Около 20% от децата с тип 2 проявяват симптоми между 3 и 15 месеца.
Детето може:
- Да се изправи в даден момент (може да се забави)
- Да не може да стои или да ходи без помощ
- Да има проблеми с преглъщането
- Да има проблеми с дишането
- Да има изкривен гръбнак (сколиоза)
- Да има ограничено движение на ставите
- Да получи проблеми със срастването на долната челюст
Детето може да загуби способността си да седи, докато стане тийнейджър. Почти две трети от децата със SMA тип 2 живеят до около 25-годишна възраст.
Тип 3 (хронична форма, започваща в детството)
Около 3 от всеки 10 души с тип 3 проявяват симптоми между 18 месеца и зряла възраст. Мускулната слабост може да означава, че:
- Имате проблеми с бягането
- Имате проблеми с изкачването на стълби
- Може да падате по-често
- Нуждаете се от помощ при ставане от стол
- Може да се нуждаете от инвалидна количка, за да се придвижвате, когато пораснете
- Имате проблеми с краката
- Вероятно няма да имате проблеми с дишането
- Вероятно нямате сколиоза (изкривен гръбнак)
Тип 4 (най-леката и най-рядка форма, започваща в зряла възраст)
Около 1 на всеки 20 души със СМА има тип 4. Симптомите започват около 30-годишна възраст:
- Може да ходите и никога да не се нуждаете от инвалидна количка
- Имате мускулна слабост в долната част на краката и крайниците
- Имате потрепвания
- Нямате проблеми с дишането
Физиотерапията помага. Много хора с тип 4 продължават да работят в продължение на много години.
Скринингът спасява време – и животи
Ранната диагноза позволява лечението да започне по-рано, когато е доказано, че е най-ефективно, което води до значително по-добри резултати в двигателното развитие и подобрена преживяемост.
Без лечение дори деца с по-леки форми могат да развият значително увреждане с течение на времето. Заболяването водеща наследствена причина за детска смърт.

Три терапии спасяват животи
Три новаторски терапии, драстично променят перспективите за хора със СМА, превръщайки опустошителната диагноза в лечимо състояние.
Благодарение на тях много деца не само оцеляват, но и постигнат ключови етапи като самостоятелно сядане, ходене и дишане без апаратна помощ, пише medicalexpress.

Генна терапия – еднократна интравенозна инфузия, която доставя функциониращо копие на SMN1 директно в моторните неврони и подобрява функциите им. Въведена е в България през 2022 г.
Модифициран антисенс олигонуклеотид – прилага се интратекално и „препрограмира“ SMN2, за да синтезира повече полезен протеин. Медикаментът е одобрен за всички форми на СМА.
Молекула-модификатор на сплайсинга на РНК – Evrysdi е името на първото и единствено перорално лекарство за СМА, което може да се приема у дома. То действа, като увеличава производството на протеин SMN в централната нервна система и в цялото тяло. одобрен за употреба при пациенти с всички видове СМА и от всички възрасти.
Какво следва в научните лаборатории?
Въпреки че съществуващите терапии трансформират лечението, изследователите продължават да рабогтят с фокус върху подобряване на резултатите през целия живот. В момента се проучват няколко обещаващи пътя:
Подобряване на доставянето и достъпа до лекарства. Полагат се усилия за максимално използване на начина, по който се прилагат настоящите лечения, като се минимизира инвазивността, намалява честотата на приемане, подобрява се тяхната трайност и се разширява достъпът по целия свят.

Регенеративни и възстановителни подходи. Учените изследват начини за възстановяване или регенериране на моторни неврони и мускулна тъкан, вече загубени от СМА. Това включва терапии със стволови клетки, както и използването на невропротективни средства и мускулно-насочени лечения. Идеята е укрепване на мускулната функция и възстановяване на мобилността при индивиди, които започват лечение по-късно в живота.
Комбинирани терапии: Тъй като нито едно лечение не обхваща напълно всеки аспект на заболяването, комбинираните стратегии се превръщат във все по-голям фокус на изследванията.
Стратегии за ранно откриване и интервенция: Изследванията са фокусирани и върху все по-ранна диагностика, дори преди раждането. Напредъкът в пренаталния скрининг, съчетан с нововъзникващите терапии в утробата, може един ден да позволи интервенция преди моторните неврони да започнат да дегенерират, предлагайки потенциал за наистина предсимптоматична грижа.
Съдържанието е информативно и не представлява консултация, препоръка или съвет. При въпроси относно вашето здраве, медицинско състояние или лечение, задължително се консултирайте с медицински специалист.
























